








Focus on Alpha Thalassemia
By Ash Lal, MD
May 2015
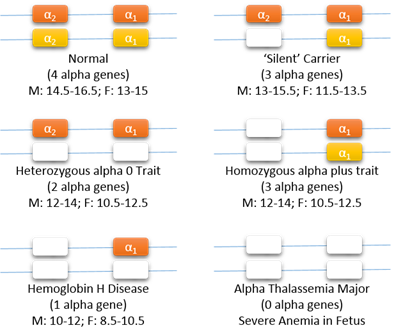
Genetic basis of alpha thalassemia:
Two alpha genes (α2 and α1) are located on each chromosome 16 (red and yellow chromosomes are inherited from different parents). White boxes indicate nonfunctioning alpha genes. The usual hemoglobin range in adults (males and females) is shown for each genotype.
A series to mark the establishment of the Multidisciplinary Center for Intrauterine Therapy in Alpha Thalassemia Major: a collaboration between UCSF Benioff Children’s Hospital Oakland and the UCSF Fetal Treatment Center.
What is Alpha Thalassemia?
Thalassemia is caused by the inability of red blood cells to synthesize hemoglobin. Every hemoglobin molecule is formed by four globin proteins (tetramers), of which two are alpha-like globins (alpha or zeta) and two are beta-like globins (beta, gamma, delta, or epsilon). Tetramers of various combinations of these globin proteins produce hemoglobins suited to different stages of life. The most common hemoglobins are adult hemoglobin, or HbA (alpha-beta), and fetal hemoglobin (alpha-gamma). Thalassemias are named after the particular globin proteins that have their synthesis affected by gene mutations. For example, in beta thalassemia, mutations in beta globin gene cause deficiency of beta globin. Similarly, there is deficiency of alpha globin due to nonfunctioning alpha globin genes in alpha thalassemia.
There is one beta gene on each chromosome 11 (one inherited from the mother and one from the father). Absence of one beta gene causes beta thalassemia trait, while absence of both genes causes beta thalassemia major. As shown in the figure, alpha genes are different, since there are two copies (called alpha 1 and alpha 2) on each chromosome 16. Most alpha thalassemia is caused by large deletions, and only some cases are the result of point mutations in alpha genes. These genetic factors are responsible for the tremendous variability in the clinical symptoms seen in alpha thalassemia. If one out of four alpha genes is absent, the hemoglobin level and size of red blood cells are slightly reduced, but there is overlap with the range in the general population. This condition is called alpha thalassemia silent carrier or heterozygous alpha plus (α+). Next, the absence of two out of four alpha genes causes a definite decrease in hemoglobin level and red blood cell size which can be detected with routine blood tests. This condition is called alpha thalassemia trait (homozygous α+ trait if the missing genes are on opposite chromosomes or heterozygous α0 trait if both missing genes are on the same chromosome). The anemia, which is mild and causes no symptoms, can be mistaken for iron deficiency anemia or beta thalassemia trait. DNA testing is needed to make a diagnosis of silent carrier and alpha thalassemia trait.
The more severe forms of alpha thalassemia are caused by the absence of either three or all four alpha genes. When three alpha globin genes are missing, patients have hemoglobin H (HbH) disease. This is associated with a moderate anemia. The common type of HbH disease does not require transfusions, but individuals with variants of HbH -- particularly HbH Constant Spring -- may need occasional transfusions. Detailed DNA analysis is essential in all of these patients, as it can predict the clinical course of the disease. HbH disease can be diagnosed during newborn screening.
Alpha Thalassemia Major
The most severe form of alpha thalassemia is alpha thalassemia major or Bart’s hydrops fetalis. This disease occurs when all four alpha genes are missing. During fetal life, the main hemoglobin is fetal hemoglobin (HbF), which is made up of alpha-gamma globin tetramers. Even earlier hemoglobins are called embryonic hemoglobins, which consist of combinations of alpha or zeta globins with epsilon or gamma globins. The importance of alpha globin during fetal life is what makes alpha thalassemia major different from beta thalassemia major. The synthesis of fetal hemoglobin (alpha-gamma) is unaffected in beta thalassemia major, so that babies are born with normal hemoglobin levels but develop anemia after several months. In alpha thalassemia, fetal hemoglobin cannot be synthesized; therefore, anemia starts before the baby is born. This profound anemia has severe effects on the development of the fetus, and alpha thalassemia major is fatal during fetal life or soon after birth. The term hydrops refers to the consequences of anemia, which causes massive enlargement of liver and spleen and, eventually, heart failure. Survival is only possible if blood transfusions are given to the fetus (intrauterine transfusions) and then continued after birth throughout life. There is also an increased risk of maternal complications when the fetus has hydrops.
Alpha thalassemia major is more common in individuals of Southeast Asian and Chinese origin. It can be successfully treated provided the pregnancy is anticipated or the diagnosis is made early with ultrasound and genetic testing of the fetus. Several patients have been cured with bone marrow transplants after birth.
The establishment of the Multidisciplinary Center for Intrauterine Therapy in Alpha Thalassemia Major brings together expertise in diagnostic, prenatal, intrauterine, perinatal, and hematological management for this complex disorder. The center is a collaboration between the UCSF Benioff Children’s Hospital Oakland Thalassemia Program and the UCSF Fetal Treatment Center.
Physicians and families who would like more information about our program and treatment or consultation for their families, please contact:
Dr. Elliott Vichinsky
(510) 428-3651 or EVichinsky@mail.cho.org
Northern California Comprehensive Thalassemia Center: (510) 428-3347
Dr. Tippi MacKenzie
tippi.mackenzie@ucsfmedctr.org
UCSF Fetal Treatment Center: 1 (800) RX-FETUS.
Updated 5/26/2015



