








Alpha Thalassemia
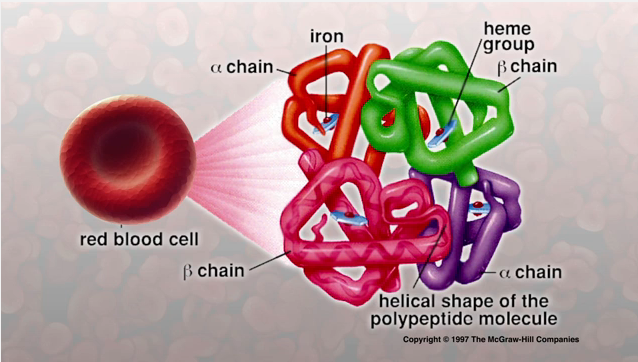
Hemoglobin Structure
from: Sylvia S. Mader, Inquiry into Life, 8th edition, McGraw-Hill, 1997.
Alpha thalassemia refers to a group of disorders characterized by inactivation of alpha globin genes. This results in a relative increase in nonfunctional beta globin or gamma globin tetramers and subsequent cell damage. Normally, there are four alpha genes. Absence or non-function of three alpha genes results in hemoglobin H disease, and the loss of all four alpha genes usually results in intrauterine death.
The alpha thalassemias are caused by a decrease in production of alpha globin chains due to a deletion or mutation of one or more of the four alpha globin genes located on chromosome 16. Alpha gene mapping can be obtained to determine the specific mutation. The alpha thalassemias can be generally categorized as: Silent Carrier, Alpha Thalassemia Trait, Hemoglobin H disease, Hemoglobin H-Constant Spring, and Alpha Thalassemia major. Frequently, the diagnosis of alpha thalassemia trait in a parent is discovered after the birth of an affected child.
The Silent Carrier status is characterized by three functional genes that code for the production of alpha globins (-α/αα). Outside the newborn period, it is not possible to make this diagnosis by conventional methods. There is overlap between the red blood cell indices of these individuals and normals, although the MCV may be slightly lower. The silent carrier will experience no health problems in his/her lifetime. This carrier state is diagnosed by deduction when a 'normal' individual has a child with Hgb H disease or with microcytic anemia consistent with alpha thalassemia trait. An unusual case of the silent carrier state is the individual who carries the Hemoglobin Constant Spring mutation [(αcsα/αα) or (ααcsαα)]. This is an elongated α-globin due to a termination codon mutation. Individuals who have this mutation have normal red blood cell indices, but can have children who have Hgb H-Constant Spring disease if the other parent has alpha thalassemia trait (--/αα). Generally, children with Hgb H-Constant Spring are more affected clinically than children who have classic Hgb H disease. Two Constant Spring carriers can also pass on their genes to have a child with Homozygous Constant Spring, a condition that has similar clinical implications as Hemoglobin H disease.
Alpha thalassemia trait is characterized by two functional genes that code for the production of alpha globins [(-α/-α) or (--/αα)]. The two genes can either occur on the same chromosome (cis-type) or on each of the pair (trans-type). Cis-type α-thalassemia trait tends to be found in individuals of Asian descent, while trans-type tends to run in individuals of African descent. Cis-type can be co-inherited with another cis-type or hemoglobin H disease to result in alpha thalassemia major, or hydrops fetalis. Individuals who have alpha thalassemia trait are identified by microcytosis, erythrocytosis, hypochromia, and mild anemia. The diagnosis is made by a combination of family studies and the ruling out of both iron deficiency anemia and beta thalassemia trait. In the neonatal period, when hemoglobin Bart's (γ4) is present, the diagnosis can be strongly suspected. In children, there are no markers such as Hgb A2 and Hgb F to make the diagnosis. (One exception is the case where both of the deletions occur on the same chromosome and zeta [ζ] globin is expressed in carriers. This is most common in Southeast Asians.) The diagnosis is one of exclusion. The clinician should be satisfied with the presumed diagnosis if the above criteria are met. During pregnancy, the microcytic anemia can be mistaken for anemia of pregnancy.
The individual with α thalassemia trait will experience no significant health problems except a possible slight anemia which cannot be treated with iron.
Hemoglobin H disease is characterized by one functional gene that codes for the production of alpha globins (--/-α). Hgb H disease should be considered in the case of a neonate in whom all of the red blood cells are very hypochromic. These neonates have a high percentage of hemoglobin Bart's on the their newborn screening results. In older children, this hemoglobinopathy is characterized by moderate anemia with a hemoglobin in the 8 to 10 gm/dL range, hypochromia, microcytosis, red cell fragmentation, and a fast migrating hemoglobin (Hgb H) on electrophoresis.
Hemoglobin H does not function as a normal hemoglobin and has a high oxygen affinity (holds onto oxygen longer making it unavaible for use by the body), so the measured hemoglobin in these children is misleading. Individuals who have Hgb H generally have a persistent stable state of anemia, which may be accentuated by increased hemolysis during viral infections and by exposure to oxidant medications, chemicals and foods such as sulfa drugs, benzene, and fava beans (similar to individuals who have G6PD deficiency). As the red cells mature they loose their ability to withstand oxidant stress and Hgb H precipitates, leading to hemolysis. Therapy for individuals who have Hgb H disease includes folate, avoidance of oxidant drugs and foods, genetic counseling education and frequent medical care. Uncommon occurrences in a child with Hgb H would be severe anemia, skin ulceration, and splenomegaly requiring splenectomy. Unlike individuals who have beta thalassemia, hemosiderosis is rare in Hgb H disease.
Children with Hemoglobin H-Constant Spring (--/αcsα) have a more severe course than children who have Hgb H. They have a more severe anemia, with a steady state hemoglobin ranging between 7 and 8 gm/dl. They more frequently have splenomegaly and severe anemia with febrile illnesses and viral infections, often requiring transfusion. If anemia is chronically severe and the child has splenomegaly, a splenectomy may be performed. If splenectomy is anticipated, the complication of severe post-splenectomy thrombocytosis with hypercoagulability can occur, leading to thrombosis of the splenic vein or hepatic veins. This complication has also been reported as recurrent pulmonary emboli and clotting diathesis. At UCSF Benioff Children's Hospital Oakland, children who are scheduled to have surgery are treated pre-surgically with low molecular weight heparin, followed by low dose aspirin, continued indefinitely.
The most severe form of alpha thalassemia is Alpha Thalassemia Major or hydrops fetalis, characterized by a deletion of all four genes that code for alpha globins (--/--). This diagnosis is frequently made in the last months of pregnancy when fetal ultrasound indicates a hydropic fetus. The mother frequently exhibits toxemia and can develop severe postpartum hemorrhage. These infants are usually stillborn. There can be other congenital anomalies, though none are pathognomonic for alpha thalassemia major. Since alpha globins are required for production of fetal and adult hemoglobin, the fetus suffers from signifiant in utero hypoxia. The only hemoglobins found in these infants are: Hgb Portland (ζ2γ2), Hgb H (β4), and Hgb Bart's (γ4), and no Hgb A or A2. These babies can have other complications associated with hydrops, such as heart failure and pulmonary edema.
If the diagnosis is made early, intrauterine transfusions can be performed. There are reports of survival and chronic transfusion in these infants; CHO cares for one alpha thalassemia major baby with chronic transfusion therapy. Undoubtedly, more of these infants could be saved if the diagnosis is anticipated by prenatal diagnosis and treatment provided.



