






![]()
 The Center for Maternal-Fetal Precision Medicine is a trans-disciplinary program designed to improve our understanding and treatment of patients with congenital anomalies and pregnancy complications. We aim to integrate resources and talent between scientists and clinicians to accelerate research and clinical trials of fetal and neonatal interventions,
create stronger bridges between basic research and clinical applications, and improve maternal, fetal, and neonatal care.
The Center for Maternal-Fetal Precision Medicine is a trans-disciplinary program designed to improve our understanding and treatment of patients with congenital anomalies and pregnancy complications. We aim to integrate resources and talent between scientists and clinicians to accelerate research and clinical trials of fetal and neonatal interventions,
create stronger bridges between basic research and clinical applications, and improve maternal, fetal, and neonatal care.
Intrauterine Therapy for Alpha Thalassemia Major:
a Multidisciplinary Center
 Fetal Transplant Study
Fetal Transplant Study
UCSF Benioff Children's Hospital Oakland and the Center for Maternal-Fetal Precision Medicine have begun enrollment for a clinical trial
that will test the safety of combining in utero hematopoietic stem cell transplant with a fetal transfusion of red blood cells. This combination is
aimed at treating and, possibly, curing Alpha Thalassemia Major, a blood disease that is often fatal in utero. This trial is the first of its kind
in the world, and could also lead to treatments for other life-threatening blood diseases, such as sickle-cell anemia.
...read more
Background
The UCSF Fetal Treatment Center and UCSF Benioff Children's Hospital Oakland Thalassemia Center have established the first multidisciplinary center for Alpha Thalassemia Major. The program is designed to address the complex diagnostic, prenatal, intrauterine, and perinatal management issues affecting a family with an Alpha Thalassemia Major pregnancy. Maternal complications of an Alpha thalassemia pregnancy are common and can be serious. Our perinatal program is designed to monitor these pregnancies in order to prevent or minimize these risks. Families at risk for an Alpha thalassemia pregnancy are confronted with significant medical, psychological, economic and ethical issues which are addressed by our multidisciplinary team. Fetal therapy, including intrauterine transfusion, is relatively safe when performed by experienced perinatologists, and dramatically improves the likelihood of fetal survival. Families undergo education concerning the risks and benefits of intervention. As more babies are being born with Alpha Thalassemia Major, updated information on the long-term prognosis and quality-of-life are shared with potential parents.
Alpha Thalassemia Major: Clinical and Laboratory Picture
Hemoglobin Bart's (Hydrops Fetalis or Alpha Thalassemia Major) is a devastating, usually fatal disease. It is common in many ethnic groups, including China, Southeast Asia, the Philipines, Greece, Turkey, Cyprus, India, Sardinia, and many other parts of the world. In the first 8 weeks of gestation, embryonic hemoglobin effectively carries oxygen to the tissues. Alpha Thalassemia Major due to lack of Alpha chains, prevents the fetus from producing fetal hemoglobin and therefore, oxygen cannot be effectively delivered to the tissues. As the fetus becomes progressively anemic, massive enlargement of the liver and spleen occur in an unsuccessful attempt to produce more red cells. Eventually, the fetus develops heart failure, severe body edema, and intrauterine demise often follows.
Maternal Complications
Serious maternal complications can occur in women during an Alpha Thalassemia Major pregnancy. These are high-risk pregnancies that require a multidisciplinary perinatal team. Patients often experience hypertension, pre-eclampsia, and are at risk for hemorrhage, anemia, infections, renal failure, premature labor, congestive heart failure, abruptio placenta and oligohydramios. It is critical that women with Alpha Thalassemia Major pregnancies receive high-risk perinatal care.
Perinatal Treatment: Intrauterine Transfusion and Stem Cell Transplantation
The Impact of Early Detection and Intrauterine Transfusion Therapy on Growth and Neurocognitive Function
Early detection of fetal anemia allows for family planning and the option for intrauterine intervention. Doppler ultrasound of the fetal cerebral artery circulation is a very sensitive and specific test for predicting anemia. In addition, measuring fetal blood flow and other fetal changes including placental thickness can detect Alpha Thalassemia Major by as early as 12 weeks of age, before the development of fetal and maternal complications. Serial prenatal ultrasonography starting by 14 weeks of gestation will detect the development of anemia and allow for intrauterine transfusions, which correct the fetal anemia and usually prevents fetal loss. Long-term follow-up of survivors of severe fetal anemia suggest that neurologic function is maintained and childhood quality of life appears good. Childhood follow-up studies at 5 years of age for Alpha Thalassemia Major and other pregnancies associated with severe fetal anemia are limited but very encouraging. Follow-up of approximately 30 children have normal neurocognitive and motor function, and almost all are attending standard school educational programs. Neurocognitive testing on 16 children who had hydrops fetalis and underwent intrauterine transfusion indicate normal IQ testing in most subscales.
Our team is also investigating the role of in utero transplantation for fetuses with Alpha thalassemia. The main advantage of this strategy is to take advantage of the unique fetal immune system so that the fetus could become tolerant to the transplanted cells. Such an approach may avoid the toxicity that is associated with routine bone marrow transplantation. Evidence from animal models supports the use of this strategy in select hematopoietic diseases and our team is investigating techniques to improve the success of in utero transplantation prior to initiating a clinical trial.
Basic information on hemoglobin and Alpha thalassemia
The hemoglobin is the molecule within the red cell that carries oxygen to all the body's tissues. It is essential for life. The type of hemoglobin present in the red cell changes during prenatal life. Embryonic hemoglobin is the main hemoglobin in the first few months of life. After 8 weeks of gestation, fetal hemoglobin rapidly increases and replaces embryonic hemoglobin -- until after birth when it is replaced by adult hemoglobin.
All hemoglobin (except for embryonic hemoglobin) consists of 4 globin chains, which always contain two Alpha Chains and two non-Alpha Chains. Fetal hemoglobin is made up of 2 Alpha chains and 2 gamma chains. Adult hemoglobin (Hemoglobin A) is made up of 2 Alpha and 2 Beta chains. This means that a mutation in the Alpha chain will affect all hemoglobin production after about 8 weeks of life. The Alpha globin chains are controlled by 4 Alpha globin genes, with 2 Alpha globin genes inherited from each parent. Alpha gene mapping can be obtained to determine the specific mutation. Alpha thalassemias can be categorized by the number of mutated genes.
• Silent Carrier: one mutation. Characterized by three functional genes that code for the production of Alpha globins. Outside the newborn period, it is not possible to make this diagnosis by conventional methods. These individuals have no abnormalities on their blood tests and are detected only by special tests, such as DNA analysis.
• Alpha Thalassemia Trait: two mutations. Up to 5% of the world's population has Alpha thalassemia trait. It is mild and may cause the red cell to be small in size, with slight anemia.
• Hemoglobin H disease: 3 mutations. It can cause moderate anemia and some medical problems.
• Alpha Thalassemia Major (also called Hemoglobin Bart's or Hydrops Fetalis): 4 mutations. All 4 Alpha genes are affected.
There is marked variability in the intrauterine clinical course of Alpha Thalassemia Major due to different mutations. There are over 126 Alpha thalassemia mutations. The large mutations may affect the embryonic hemoglobin essential for survival in the first few months of life. These severe mutations result in early gestational miscarriage or abortion and may go undetected. Smaller mutations do not involve the embryonic gene and may result in fetal disease developing later in gestation. Most of these pregnancies are 3rd-trimester miscarriages, stillbirths and occasionally critically ill surviving newborns. While the severity of the Alpha globin mutation is the most major factor in disease severity, there are other important genetic mutations that influence the disease severity. Sometimes there are mutations on other parts of the chromosome that affect Alpha globin gene function. Occasionally, these non-Alpha-thalassemia mutations will cause a fetus with only 3 genes affected to clinically be as severe as a fetus with 4 affected genes. Additionally, there are non-genetic factors that may aggravate fetal anemia such as ABO/Rh incompatibility in the parents. There are increasing numbers of surviving Alpha thalassemia newborns being reported who have not been prenatally diagnosed. However, these surviving newborns usually have experienced severe fetal hypoxia due to anemia in the third trimester; they often have consequences including neurologic injury and developmental abnormalities such as skeletal malformations. The associated neurologic injury and developmental abnormalities are secondary to the severe anemia, since they are generally prevented by early intrauterine transfusion. Those who do survive the neonatal period continue to have chronic anemia and require monthly transfusion therapy and treatment for iron accumulation. Stem cell transplantation is now being successfully reported in some survivors.
Post-Natal Treatment: Multidisciplinary Care with Chronic Transfusions
Following birth, Alpha Thalassemia Major patients require monthly transfusion therapy and medication to prevent iron accumulation. This requires care in a multidisciplinary thalassemia program. Access to new oral iron chelators and specialized equipment to monitor tissue iron and organ dysfunction are essential. Without adequate care, patients are at risk for premature death and multiple complications including heart failure, diabetes, growth failure, and bone disease. Patients receiving optimal care are now living into the sixth decade.
Ethical and Psychological Burdens for Families Facing a Alpha Thalassemia Major Pregnancy
The standard medical approach for an Alpha Thalassemia Major pregnancy is termination and non-support. The prognosis for Beta Thalassemia Major has dramatically changed in the last decade. Beta Thalassemia Major is a defect in the Beta gene and in contrast to Alpha Thalassemia Major, does not become symptomatic until after birth. The standard therapy for these infants are chronic monthly transfusions and medication to prevent iron overload. The availability of safe blood and oral iron chelation therapy has resulted in many Beta Thalassemia Major patients living a productive life throughout adulthood. These results have increased the interest in intrauterine therapy for Alpha Thalassemia Major pregnancies. Each family requires objective information in a supportive environment that respects the parental attitudes and aids in their decision analysis. Follow-up support following the family's decision is an important aspect of care.
Recommendations and Services for At-Risk Couples
 This is a video made by a wonderful family who understood the issues surrounding Alpha Thalassemia Major pregnancies who were committed to doing everything they could to help their child. It is a moving life story of their baby's first year and illustrates the family's challenges and successes. We are posting this with the family's permission because they want this information about Alpha Thalassemia Major to be available to the community.
This is a video made by a wonderful family who understood the issues surrounding Alpha Thalassemia Major pregnancies who were committed to doing everything they could to help their child. It is a moving life story of their baby's first year and illustrates the family's challenges and successes. We are posting this with the family's permission because they want this information about Alpha Thalassemia Major to be available to the community.
Prenatal Testing of Parents
Alpha Thalassemia Major or Bart's Hydrops occurs when both parents are carriers for Alpha thalassemia. Since Alpha thalassemia is an autosomal recessive condition, both parents are carriers for thalassemia (heterozygotes). When both parents are carriers, there is a 25% chance that the pregnancy will have Alpha Thalassemia Major. Up to five percent of the population may be carriers, particularly in high-risk ethnic groups. Most parents have not been tested for Alpha thalassemia. Alpha thalassemia trait is very mild, and may be missed by routine blood tests. Hemoglobin level and hemoglobin electrophoresis are often normal in people who are carriers. Microcytosis (small red cells) may be seen but has many different etiologies and may be in the normal range. In general, the MCV (mean corpuscular volume) is below 82 in people who are carriers, but this is an unreliable, non-definitive test for Alpha thalassemia. MCH below 27 pg is suggestive of thalassemia trait.
Molecular diagnosis of both parents is necessary to accurately determine Alpha thalassemia status of the fetus. The mother should be tested first. If she has abnormalities, then testing of the father is necessary. There are over 50 different mutations for Alpha thalassemia. It is optimal that at-risk couples are tested for their Alpha thalassemia status before pregnancy occurs, but testing is always performed as part of the evaluation of a presumed Alpha thalassemia pregnancy.
If the mother has microcytosis without iron deficiency, DNA diagnosis of thalassemia is recommended. Sample Testing instructions can be found on the Hemoglobinopathy Laboratory web site.
Fetal Diagnosis
When parents have Alpha thalassemia trait, DNA analysis of the fetus is required. Fetal tissue obtained by chorionic villus sampling early in the first trimester is indicated. This is usually performed at 10 to 12 weeks of gestation. Alternatively, cultured cells from amniotic fluid obtained by amniocentesis may be done at 15 weeks gestation. Non-invasive prenatal diagnosis is being developed utilizing purified fetal DNA from a simple maternal blood sample is being studied. This is a research test because of the problem of maternal DNA interfering with the fetal testing.
Fetal diagnosis by Doppler ultrasonography
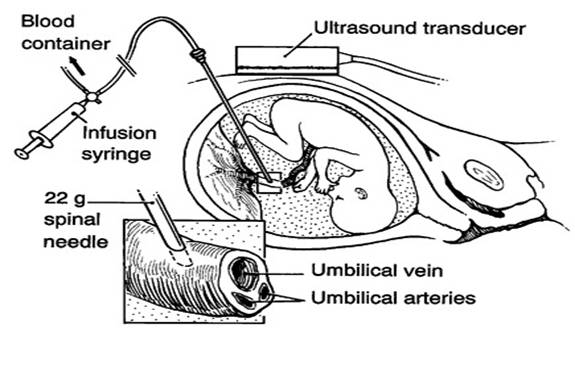 Intrauterine Transfusion
Intrauterine Transfusion
Doppler ultrasonography over the mother's abdomen can determine the blood flow in the cerebral arteries of the fetus. The blood flow rate strongly correlates with anemia in the fetus. Greater than 90% of cases of Alpha Thalassemia Major or a severe anemia can be detected safely with this Doppler technique. Middle cerebral artery measurements can be monitored after 16 weeks of gestation and reliably determine if severe fetal anemia exists.
Intrauterine transfusion
Intrauterine transfusion is a relatively safe procedure when performed by perinatologists familiar with the technique. However, there are risks that include a 1% fetal death rate and 5% chance of usually mild bradycardia; overall, the serious complication rate is approximately 3%. Intrauterine transfusion and testing of the fetal blood can be done through several techniques. Access through the umbilical cord is commonly used. The sample is tested for severe anemia and the diagnosis of Alpha thalassemia. The degree of anemia and the appearance of the cells are usually immediately diagnostic. DNA testing is always indicated for 100% certainty. A fetal hemoglobin below 7 gm/dl is severe and consistent with Alpha Thalassemia Major. Following acquisition of the fetal sample, an intrauterine transfusion is performed. Fresh blood that is CMV-negative, leuko-depleted, washed and irradiated is used. The units are washed in order to increase the hemoglobin concentration. Serial transfusions are often necessary. The correction of the anemia usually results in a dramatic improvement in fetal function, and a correction of the cerebral blood flow rate. With appropriate intrauterine transfusion, almost 90% of these pregnancies result in a live birth.
Perinatal Delivery of an Alpha Thalassemia Major Baby
The timing of the delivery is related to allowing the fetus to mature as much as possible. However, most fetal births of Alpha Thalassemia Major babies initially are unstable and may develop respiratory distress. Many may require temporary ventilation support in order to adjust to post-natal life. Despite the relatively high rate of transient neonatal problems, most of these babies fully recover. The fetal blood cells (Bart's hemoglobin) in Alpha Thalassemia Major that circulate in the newborn do not carry oxygen effectively. Therefore, an exchange transfusion at birth is often required. After the baby stabilizes in the neonatal period, s/he requires a monthly transfusion in order to sustain a healthy hemoglobin level. These transfusions can be given as an outpatient. Ongoing monitoring of Bart's hemoglobin is necessary to make sure it does not become prevalent. Transfusions suppress its production.
Chronic Transfusion Therapy
Once discharged, the babies are transfused close to their homes as an outpatient, indefinitely. After a year of age, medications to protect them from the excess iron found in blood are started.
Long-term Neurologic Prognosis
Serious neuro-developmental impairment is found in 5% of these babies: this may include significant developmental delay, cerebral palsy, deafness, and rarely, blindness. While other children may have mild neurologic impairment, most function well and are mainstream in school. The degree of impairment is influenced by how severely affected the fetus was before intrauterine transfusion therapy was initiated.
Curing Alpha Thalassemia After Birth

Stem Cell Transplantation
Recent advances in stem cell transplantation have resulted in some patients being cured. Successful cases of related, unrelated, and mismatched stem cell transplantation for Alpha Thalassemia Major are now possible. Advances in in-utero therapy have resulted in long-term survival, precipitating studies of long-term survivors.
Alpha Thalassemia Major survivors can grow up and live a productive life. The burden of chronic transfusion therapy and its complications have resulted in pilot studies evaluating curative stem cell treatment. Several infants with Alpha Thalassemia Major have been cured with a stem cell transplantation from a sibling. This success has led to transplantation studies utilizing genetically-matched, unrelated donors. Though experimental, studies evaluating their success are increasing. At UCSF Benioff Children's Hospital Oakland, we follow several children who have undergone curative transplantation for thalassemia, including a teenager who had successful transplantation for Alpha Thalassemia Major as an infant.
Experimental Therapy to Cure Alpha Thalassemia In-Utero with Gene Therapy
Gene therapy trials have now been initiated in thalassemia. In the near future, the Fetal Medicine Program and the Thalassemia Center are investigating the potential for a treatment utilizing in-utero gene therapy. While there are several obstacles to the success of such a program, its development is progressing.
Our Staff
-
Elliott Vichinsky, MD:
Medical Director, Hematology/Oncology
 Dr. Vichinsky is the Director of Hematology/Oncology at UCSF Benioff Children's Hospital Oakland, an Adjunct Professor at University of California San Francisco, as well as the Director of the Northern
California Sickle Cell and Thalassemia Centers. He established the hemoglobinopathy center over 30 years ago at Children's
Hospital Oakland to provide primary through tertiary care to pediatric and adult patients. He is a board-certified pediatric hematologist/oncologist whose major interest is in understanding and improving the care of patients with hemoglobinopathies. His career has focused on translational research in hemoglobinopathies to answer therapeutic questions in hematologic and iron overload disorders. He has been a federally-funded research investigator for over 30 years and been national principal investigator for many successful trials -- including transfusion therapy and chelation therapy in hemoglobinopathies.
He has been principal investigator or co-investigator on many of the key translational studies, including understanding the mechanism and treatment of neurologic injury in sickle cell disease; iron trafficking and iron overload in hemoglobinopathies, and new therapies to change the pathophysiology of sickle cell biology. He has served as Editor-in-Chief of Pediatric Hematology and Oncology; Chairman of the Thalassemia Clinical Research Network, and Director of the Cooley’s Medical Board. Dr. Vichinsky has published over 300 articles and several books, and has received lifetime achievement awards from the Cooley’s Anemia Foundation and the National Sickle Cell Disease Association.
Dr. Vichinsky is the Director of Hematology/Oncology at UCSF Benioff Children's Hospital Oakland, an Adjunct Professor at University of California San Francisco, as well as the Director of the Northern
California Sickle Cell and Thalassemia Centers. He established the hemoglobinopathy center over 30 years ago at Children's
Hospital Oakland to provide primary through tertiary care to pediatric and adult patients. He is a board-certified pediatric hematologist/oncologist whose major interest is in understanding and improving the care of patients with hemoglobinopathies. His career has focused on translational research in hemoglobinopathies to answer therapeutic questions in hematologic and iron overload disorders. He has been a federally-funded research investigator for over 30 years and been national principal investigator for many successful trials -- including transfusion therapy and chelation therapy in hemoglobinopathies.
He has been principal investigator or co-investigator on many of the key translational studies, including understanding the mechanism and treatment of neurologic injury in sickle cell disease; iron trafficking and iron overload in hemoglobinopathies, and new therapies to change the pathophysiology of sickle cell biology. He has served as Editor-in-Chief of Pediatric Hematology and Oncology; Chairman of the Thalassemia Clinical Research Network, and Director of the Cooley’s Medical Board. Dr. Vichinsky has published over 300 articles and several books, and has received lifetime achievement awards from the Cooley’s Anemia Foundation and the National Sickle Cell Disease Association.
-
Julian Parer, MD, PhD:
UCSF Professor, Perinatology
 Dr. Julian Parer is a perinatologist in UCSF Women's Health at Parnassus. He specializes in maternal-fetal medicine and high-risk pregnancies and has extensive experience caring for pregnant women with pregnancy loss and premature delivery, medical illnesses and Rh disease as well as most other complications of pregnancy. In addition, he works with the Fetal Treatment Center at UCSF Benioff Children's Hospital. Together with all members of the obstetrical team, he is committed to enabling all of his patients to have as positive a pregnancy and birth experience as possible.
Dr. Parer earned a medical degree at the University of Washington in Seattle and completed a residency at the University of Southern California. He is board certified in obstetrics and gynecology as well as maternal-fetal medicine.
Dr. Julian Parer is a perinatologist in UCSF Women's Health at Parnassus. He specializes in maternal-fetal medicine and high-risk pregnancies and has extensive experience caring for pregnant women with pregnancy loss and premature delivery, medical illnesses and Rh disease as well as most other complications of pregnancy. In addition, he works with the Fetal Treatment Center at UCSF Benioff Children's Hospital. Together with all members of the obstetrical team, he is committed to enabling all of his patients to have as positive a pregnancy and birth experience as possible.
Dr. Parer earned a medical degree at the University of Washington in Seattle and completed a residency at the University of Southern California. He is board certified in obstetrics and gynecology as well as maternal-fetal medicine.
-
Mark C. Walters, MD:
Director, Blood and Bone Marrow Transplant Program
 Dr. Walters is the Jordan Family Director of the Blood and Marrow Transplantation Program at UCSF Benioff Children's Hospital Oakland and Associate Adjunct Professor in the Department of Pediatrics at the
University of California, San Francisco School of Medicine. Dr. Walters received his A.B. with honors in
Genetics from the University of California, Berkeley and his MD from the University of California, San Diego.
He completed pediatric residency training at the University of Washington and hematology/oncology fellowship
training at the University of Washington and the Fred Hutchinson Cancer Research Center in Seattle.
He was a junior faculty member in Seattle before matriculating to Oakland in 1999. He has been active
in cooperative clinical transplantation trials and has led several NIH-supported investigations of
hematopoietic cell transplantation for sickle cell anemia and thalassemia. He has authored or
co-authored many publications with a focus on hematopoietic cell transplantation for hemoglobin
disorders, and he has a research interest in the application of umbilical cord blood transplantation
and other novel cellular therapies for hereditary hematological disorders.
Dr. Walters is the Jordan Family Director of the Blood and Marrow Transplantation Program at UCSF Benioff Children's Hospital Oakland and Associate Adjunct Professor in the Department of Pediatrics at the
University of California, San Francisco School of Medicine. Dr. Walters received his A.B. with honors in
Genetics from the University of California, Berkeley and his MD from the University of California, San Diego.
He completed pediatric residency training at the University of Washington and hematology/oncology fellowship
training at the University of Washington and the Fred Hutchinson Cancer Research Center in Seattle.
He was a junior faculty member in Seattle before matriculating to Oakland in 1999. He has been active
in cooperative clinical transplantation trials and has led several NIH-supported investigations of
hematopoietic cell transplantation for sickle cell anemia and thalassemia. He has authored or
co-authored many publications with a focus on hematopoietic cell transplantation for hemoglobin
disorders, and he has a research interest in the application of umbilical cord blood transplantation
and other novel cellular therapies for hereditary hematological disorders.
-
Tippi C. MacKenzie, MD:
Associate Professor of Surgery and Director of Research, UCSF Fetal Treatment Center
 Tippi C. MacKenzie, MD is Associate Professor of Surgery and the Director of Research in the UCSF Fetal Treatment Center. She graduated from Harvard and Stanford Medical School prior to her surgical training at Brigham and Women’s Hospital and The Children’s Hospital of Philadelphia. As a pediatric/fetal surgeon, her primary goal is to apply stem cell therapies to treat fetuses with congenital anomalies. Dr. MacKenzie has an active laboratory, the The MacKenzie Lab, that studies the mechanisms of tolerance induction following in utero stem cell transplantation. Her team has helped to establish that the maternal immune response is an important barrier to engraftment and is also testing additional strategies to create space in the fetal hematopoietic niche. Since many fetuses with Alpha thalassemia also develop hydrops, she is also investigating the mechanisms that lead to hydrops and its downstream effects, such as preterm labor. Her work has been supported by the NIH, the March of Dimes, and the California Institute for Regeneration Medicine.
Tippi C. MacKenzie, MD is Associate Professor of Surgery and the Director of Research in the UCSF Fetal Treatment Center. She graduated from Harvard and Stanford Medical School prior to her surgical training at Brigham and Women’s Hospital and The Children’s Hospital of Philadelphia. As a pediatric/fetal surgeon, her primary goal is to apply stem cell therapies to treat fetuses with congenital anomalies. Dr. MacKenzie has an active laboratory, the The MacKenzie Lab, that studies the mechanisms of tolerance induction following in utero stem cell transplantation. Her team has helped to establish that the maternal immune response is an important barrier to engraftment and is also testing additional strategies to create space in the fetal hematopoietic niche. Since many fetuses with Alpha thalassemia also develop hydrops, she is also investigating the mechanisms that lead to hydrops and its downstream effects, such as preterm labor. Her work has been supported by the NIH, the March of Dimes, and the California Institute for Regeneration Medicine.
-
Ashutosh Lal, MD:
Director, Thalassemia Clinical Program
 Dr. Lal organizes the day-to-day management of patients attending our Comprehensive Thalassemia Center, and
supervises the clinical program. The Center's major focus is health maintenance of transfusion-dependent patients
by rigorous monitoring of clinical indicators, such as iron burden, cardiac function, endocrine testing, growth
and nutrition. In order to improve coordination of care for our patients living outside California, we are
designing innovative methods to communicate with their providers. Dr. Lal's research areas are the natural history
of hemoglobin H disease, developing intensive iron chelation regimens, and nutritional deficiencies in thalassemia.
Simultaneously, in the laboratory he and his team are trying to define whether iron-induced injury to mitochondria
can help to explain organ damage in thalassemia.
Dr. Lal organizes the day-to-day management of patients attending our Comprehensive Thalassemia Center, and
supervises the clinical program. The Center's major focus is health maintenance of transfusion-dependent patients
by rigorous monitoring of clinical indicators, such as iron burden, cardiac function, endocrine testing, growth
and nutrition. In order to improve coordination of care for our patients living outside California, we are
designing innovative methods to communicate with their providers. Dr. Lal's research areas are the natural history
of hemoglobin H disease, developing intensive iron chelation regimens, and nutritional deficiencies in thalassemia.
Simultaneously, in the laboratory he and his team are trying to define whether iron-induced injury to mitochondria
can help to explain organ damage in thalassemia.
-
Michael Harrison, MD:
Director Emeritus, UCSF Fetal Treatment Center; Professor of Surgery & Pediatrics
 Dr. Michael Harrison graduated cum laude from Yale University and magna cum laude from Harvard Medical School. He completed his surgical training at the Massachusetts General Hospital in Boston and his pediatric surgery fellowship at Children's Hospital of Los Angeles. He is board certified in Surgery, Pediatric Surgery, and Critical Care.
Dr. Harrison and his surgery associates confine their surgical practice exclusively to children with special interest in fetal surgery, in repair of complex birth defects involving the chest, lung, abdomen, bowel, and bladder, and surgical care of children from birth through adolescence. Dr. Harrison and his associates, The Bay Area Pediatric Surgeons, do consultations and provide surgical care at Moffitt/Long Hospitals UCSF, California Pacific Medical Center, and Kaiser Permanente, San Francisco.
Dr. Harrison has a special interest in fetal surgery which he has pioneered with his colleagues at UCSF. For the past 18 years Dr. Harrison has studied the pathophysiology and natural history of a number of life-threatening fetal abnormalities including congenital diaphragmatic hernia, congenital cystic adenomatoid malformation of the lung, sacrococcygeal teratoma, fetal obstructive uropathy, and myelomeningocele. He has developed techniques for both in-utero open fetal surgery and endoscopic surgical repair (FETENDO Fetal Surgery)of many fetal abnormalities. In addition, Dr. Harrison's team has developed in-utero stem cell transplantation to treat immunodeficiencies, enzyme deficiencies, and hemoglobinopathies. Dr. Harrison leads the multidisciplinary UCSF Fetal Treatment Center Team that has developed an international reputation for treating complex birth defects before and after birth.
Dr. Michael Harrison graduated cum laude from Yale University and magna cum laude from Harvard Medical School. He completed his surgical training at the Massachusetts General Hospital in Boston and his pediatric surgery fellowship at Children's Hospital of Los Angeles. He is board certified in Surgery, Pediatric Surgery, and Critical Care.
Dr. Harrison and his surgery associates confine their surgical practice exclusively to children with special interest in fetal surgery, in repair of complex birth defects involving the chest, lung, abdomen, bowel, and bladder, and surgical care of children from birth through adolescence. Dr. Harrison and his associates, The Bay Area Pediatric Surgeons, do consultations and provide surgical care at Moffitt/Long Hospitals UCSF, California Pacific Medical Center, and Kaiser Permanente, San Francisco.
Dr. Harrison has a special interest in fetal surgery which he has pioneered with his colleagues at UCSF. For the past 18 years Dr. Harrison has studied the pathophysiology and natural history of a number of life-threatening fetal abnormalities including congenital diaphragmatic hernia, congenital cystic adenomatoid malformation of the lung, sacrococcygeal teratoma, fetal obstructive uropathy, and myelomeningocele. He has developed techniques for both in-utero open fetal surgery and endoscopic surgical repair (FETENDO Fetal Surgery)of many fetal abnormalities. In addition, Dr. Harrison's team has developed in-utero stem cell transplantation to treat immunodeficiencies, enzyme deficiencies, and hemoglobinopathies. Dr. Harrison leads the multidisciplinary UCSF Fetal Treatment Center Team that has developed an international reputation for treating complex birth defects before and after birth.
-
James Huang, MD:
Director of Pediatric Hematology at UCSF, Co-director of the Hemophilia Treatment Center at UCSF Benioff Children's Hospital
 Dr. James Huang is an expert in treating children with blood disorders such as hemophilia, thrombophilia and hemoglobinopathies including thalassemias and sickle cell disease, as well as in addressing issues related to bone marrow failure. His research focuses on bleeding disorders, hemoglobinopathies and stem cell transplantation to treat blood disorders. He has a particular interest in Shwachman-Diamond syndrome, an inherited bone marrow failure syndrome associated with pancreatic dysfunction that also predisposes children to leukemia.
Prior to joining UCSF in 2006, Huang was a faculty member at Baylor College of Medicine and worked at Texas Children's Cancer Center and Hematology Service, where he was instrumental in starting its nationally designated hemophilia treatment center. A graduate of Harvard College, he earned a medical degree at Baylor College of Medicine. He completed a pediatrics internship and residency at Johns Hopkins Hospital and a pediatric hematology and oncology fellowship at Boston Children's Hospital and Dana-Farber Cancer Institute. At UCSF, he is an associate professor of pediatrics.
Dr. James Huang is an expert in treating children with blood disorders such as hemophilia, thrombophilia and hemoglobinopathies including thalassemias and sickle cell disease, as well as in addressing issues related to bone marrow failure. His research focuses on bleeding disorders, hemoglobinopathies and stem cell transplantation to treat blood disorders. He has a particular interest in Shwachman-Diamond syndrome, an inherited bone marrow failure syndrome associated with pancreatic dysfunction that also predisposes children to leukemia.
Prior to joining UCSF in 2006, Huang was a faculty member at Baylor College of Medicine and worked at Texas Children's Cancer Center and Hematology Service, where he was instrumental in starting its nationally designated hemophilia treatment center. A graduate of Harvard College, he earned a medical degree at Baylor College of Medicine. He completed a pediatrics internship and residency at Johns Hopkins Hospital and a pediatric hematology and oncology fellowship at Boston Children's Hospital and Dana-Farber Cancer Institute. At UCSF, he is an associate professor of pediatrics.
-
Morton Cowan, MD:
Chief of Allergy, Immunology and Blood and Marrow Transplant Division, UCSF
 Dr. Morton J. Cowan is chief of the Allergy, Immunology and Blood and Marrow Transplant Division at UCSF Benioff Children's Hospital San Francisco. He is recognized throughout the world for research in immunodeficiency diseases, the use of alternative donors and in utero stem cell transplantation. He performed the first bone marrow transplant at UCSF Medical Center for a child with severe combined immunodeficiency disease (SCID) in 1982, the first T-cell depleted transplant on the West Coast for a child with leukemia in 1985, and the first pure blood stem cell transplant from a parent to a child with SCID in North America.
Dr. Cowan earned an undergraduate degree in electrical engineering at the Massachusetts Institute of Technology and his medical degree at the University of Pennsylvania in Philadelphia. He completed an internship and residency in pediatrics and a fellowship in immunology at UCSF Medical Center. Cowan is the Principle Investigator of the Primary Immune Deficiency Treatment Consortium, which is an National Institute of Health funded research organization studying children with severe immune deficiencies representing institutions in the U.S. and Canada.
Dr. Morton J. Cowan is chief of the Allergy, Immunology and Blood and Marrow Transplant Division at UCSF Benioff Children's Hospital San Francisco. He is recognized throughout the world for research in immunodeficiency diseases, the use of alternative donors and in utero stem cell transplantation. He performed the first bone marrow transplant at UCSF Medical Center for a child with severe combined immunodeficiency disease (SCID) in 1982, the first T-cell depleted transplant on the West Coast for a child with leukemia in 1985, and the first pure blood stem cell transplant from a parent to a child with SCID in North America.
Dr. Cowan earned an undergraduate degree in electrical engineering at the Massachusetts Institute of Technology and his medical degree at the University of Pennsylvania in Philadelphia. He completed an internship and residency in pediatrics and a fellowship in immunology at UCSF Medical Center. Cowan is the Principle Investigator of the Primary Immune Deficiency Treatment Consortium, which is an National Institute of Health funded research organization studying children with severe immune deficiencies representing institutions in the U.S. and Canada.
-
Roberta Keller, MD:
Medical Director, Neonatal ECMO Program
 Dr. Roberta Keller is a neonatologist, an expert in caring for critically ill newborns, particularly those with congenital lung or heart disease. She is director of the Extracorporeal Membrane Oxygenation (ECMO) program, or artificial lung, for newborns. Her research addresses lung and heart disorders affecting newborns, such as congenital diaphragmatic hernia and patent ductus arteriosus. Her research on these conditions has been published in numerous journals.
Dr. Keller completed a medical degree, residency in pediatrics and fellowship in neonatal-perinatal medicine at UCSF, before joining the department of pediatrics faculty in 2003. She is the coordinator of the UCSF Neonatology Clinical Consensus Program and during her fellowship, she was the recipient of the Glaser Pediatric Research Network fellowship.
Dr. Roberta Keller is a neonatologist, an expert in caring for critically ill newborns, particularly those with congenital lung or heart disease. She is director of the Extracorporeal Membrane Oxygenation (ECMO) program, or artificial lung, for newborns. Her research addresses lung and heart disorders affecting newborns, such as congenital diaphragmatic hernia and patent ductus arteriosus. Her research on these conditions has been published in numerous journals.
Dr. Keller completed a medical degree, residency in pediatrics and fellowship in neonatal-perinatal medicine at UCSF, before joining the department of pediatrics faculty in 2003. She is the coordinator of the UCSF Neonatology Clinical Consensus Program and during her fellowship, she was the recipient of the Glaser Pediatric Research Network fellowship.
-
David Rowitch, MD:
Chief of Neonatology and professor of pediatrics and neurosurgery at UCSF
 Dr. David Rowitch, in addition to caring for patients, leads a laboratory that is investigating genetic factors that determine cellular development in the brain and its response to injury. Dr. Rowitch is also interested in neurological problems and brain cancer in premature infants.
Dr. Rowitch earned a medical degree at the University of California, Los Angeles and a doctorate in biochemistry at the University of Cambridge in England. He completed an internship and residency in pediatrics and a fellowship in newborn medicine at Children's Hospital in Boston. He participated in a postdoctoral fellowship at Harvard College in Cambridge. His work in neurobiology has earned him numerous awards, including the National Institute of Health Clinical Investigator Award, Kimmel Foundation Scholar Award and the James S. McDonnell Foundation Research Award. In 2008, Rowitch was appointed a Howard Hughes Medical Institute investigator.
Dr. David Rowitch, in addition to caring for patients, leads a laboratory that is investigating genetic factors that determine cellular development in the brain and its response to injury. Dr. Rowitch is also interested in neurological problems and brain cancer in premature infants.
Dr. Rowitch earned a medical degree at the University of California, Los Angeles and a doctorate in biochemistry at the University of Cambridge in England. He completed an internship and residency in pediatrics and a fellowship in newborn medicine at Children's Hospital in Boston. He participated in a postdoctoral fellowship at Harvard College in Cambridge. His work in neurobiology has earned him numerous awards, including the National Institute of Health Clinical Investigator Award, Kimmel Foundation Scholar Award and the James S. McDonnell Foundation Research Award. In 2008, Rowitch was appointed a Howard Hughes Medical Institute investigator.
-
Carolyn Hoppe, MD:
Director, Hemoglobinopathy Diagnostic Lab
 Dr. Hoppe is a hematologist/oncologist at CHO and a clinical scientist at CHORI with a focus
in translational research in sickle cell disease. Her interest in studying genetic modifiers of sickle cell
disease began early in her fellowship, while working in the laboratory of Dr. Elizabeth Trachtenberg at
CHORI. A recipient of the American Society of Hematology Fellow Scholar Award, she performed her initial studies
investigating HLA associations with stroke risk in children with sickle cell anemia. Dr. Hoppe has been a
team member of the Northern California Comprehensive Sickle Cell Program for the past 10 years. As the
medical director of the CHORI Hemoglobinopathy Laboratory and the California State Newborn Screening for
Hemoglobinopathies Follow-up Program, Dr. Hoppe has expanded the services provided by this program to
include molecular diagnostics.
Dr. Hoppe is a hematologist/oncologist at CHO and a clinical scientist at CHORI with a focus
in translational research in sickle cell disease. Her interest in studying genetic modifiers of sickle cell
disease began early in her fellowship, while working in the laboratory of Dr. Elizabeth Trachtenberg at
CHORI. A recipient of the American Society of Hematology Fellow Scholar Award, she performed her initial studies
investigating HLA associations with stroke risk in children with sickle cell anemia. Dr. Hoppe has been a
team member of the Northern California Comprehensive Sickle Cell Program for the past 10 years. As the
medical director of the CHORI Hemoglobinopathy Laboratory and the California State Newborn Screening for
Hemoglobinopathies Follow-up Program, Dr. Hoppe has expanded the services provided by this program to
include molecular diagnostics.
-
Mary Norton, MD:
Professor and Vice Chair
Clinical and Translational Genetics
 Dr. Norton’s interests include genetic testing and its application to prenatal screening and diagnosis. She focuses on the unique aspects of translating new technologies into improved care for pregnant women and their fetuses, and the complexities of the maternal fetal dyad. She leads a multinational study on the use of cell free DNA testing in average risk women for the detection of fetal aneuploidy. Current projects include mechanisms and disorders associated with nonimmune hydrops fetalis and potential for in utero therapy; and application of whole exome and genome sequencing in the prenatal context.
Dr. Norton’s interests include genetic testing and its application to prenatal screening and diagnosis. She focuses on the unique aspects of translating new technologies into improved care for pregnant women and their fetuses, and the complexities of the maternal fetal dyad. She leads a multinational study on the use of cell free DNA testing in average risk women for the detection of fetal aneuploidy. Current projects include mechanisms and disorders associated with nonimmune hydrops fetalis and potential for in utero therapy; and application of whole exome and genome sequencing in the prenatal context.
-
Juan M. González, MD, MS, FACOG:
Perinatologist; Assistant Professor, UCSF Maternal-Fetal Medicine
 Dr. Juan González is a specialist in maternal/fetal health at UCSF. He completed an internship in internal medicine at Johns Hopkins University. At the University of Pennsylvania, he completed a residency in Obstetrics and Gynecology, and a fellowship in maternal fetal medicine, along with a Masters degree in Translational Research.
Dr. Juan González is a specialist in maternal/fetal health at UCSF. He completed an internship in internal medicine at Johns Hopkins University. At the University of Pennsylvania, he completed a residency in Obstetrics and Gynecology, and a fellowship in maternal fetal medicine, along with a Masters degree in Translational Research.
-
Shannon Gaine, FNP:
Thalassemia Nurse Practitioner
 Shannon Gaine is a licensed Family Nurse Practitioner who joined the thalassemia team in May 2016.
She earned her Master’s Degree as Family Nurse Practitioner at Georgetown University.
After completing her Masters she worked at Columbia Presbyterian, New York City in the Pediatrics emergency room.
Upon moving to California, Shannon worked in a small clinic in Mountain View before coming to to UCSF Benioff Children’s Hospital Oakland.
She has an interest in working with the community to bring more awareness and education about thalassemia.
Shannon Gaine is a licensed Family Nurse Practitioner who joined the thalassemia team in May 2016.
She earned her Master’s Degree as Family Nurse Practitioner at Georgetown University.
After completing her Masters she worked at Columbia Presbyterian, New York City in the Pediatrics emergency room.
Upon moving to California, Shannon worked in a small clinic in Mountain View before coming to to UCSF Benioff Children’s Hospital Oakland.
She has an interest in working with the community to bring more awareness and education about thalassemia. -
Wendy Hyde Murphy, LCSW:
Social Worker
 Wendy is a Licensed Clinical Social Worker who joined the Thalassemia Team in August 2012 after
spending six years in the Oncology & Neuro-Oncology Department at Children’s Hospital. She earned
her Master’s Degree in Social Work from the University of Michigan. After completing her MSW,
she worked for two years in a foster care agency and then spent the next 12 years at Seneca
Center in their residential and day treatment programs for emotionally challenged children.
She was involved in direct counseling, intake coordination and supervising therapists.
Wendy is a Licensed Clinical Social Worker who joined the Thalassemia Team in August 2012 after
spending six years in the Oncology & Neuro-Oncology Department at Children’s Hospital. She earned
her Master’s Degree in Social Work from the University of Michigan. After completing her MSW,
she worked for two years in a foster care agency and then spent the next 12 years at Seneca
Center in their residential and day treatment programs for emotionally challenged children.
She was involved in direct counseling, intake coordination and supervising therapists.
-
Mahin Azimi, BS, CLS:
Medical Technologist, Hemoglobinopathy Diagnostic Lab
 Mahin Azimi is a Senior Clinical Laboratory Research Scientist and Supervisor of the CHRCO
Hemoglobinopathy Reference Laboratory. Mahin holds three Bachelor’s degrees: one in Social Sciences
from the University of California, Berkeley, one in Medical Laboratory Sciences from the
University of Geneva, Switzerland, and a Bachelor of Science in Biology with a specialty in Clinical
Laboratory Science from the California State University, Hayward. She currently holds 3 licenses in the
State of California: CLS, ASCP, NCA. She came to the laboratory at CHRCO in 1994 and has played
an integral role in building the laboratory into a nationally recognized resource for
diagnosis and interpretation of hemoglobinopathies.
Mahin Azimi is a Senior Clinical Laboratory Research Scientist and Supervisor of the CHRCO
Hemoglobinopathy Reference Laboratory. Mahin holds three Bachelor’s degrees: one in Social Sciences
from the University of California, Berkeley, one in Medical Laboratory Sciences from the
University of Geneva, Switzerland, and a Bachelor of Science in Biology with a specialty in Clinical
Laboratory Science from the California State University, Hayward. She currently holds 3 licenses in the
State of California: CLS, ASCP, NCA. She came to the laboratory at CHRCO in 1994 and has played
an integral role in building the laboratory into a nationally recognized resource for
diagnosis and interpretation of hemoglobinopathies.
Resources
![]()

- The UCSF MacKenzie Lab
- The UCSF Fetal Treatment Center. Referrals: 1-800-RX-FETUS.
- The Center for Maternal-Fetal Precision Medicine is a trans-disciplinary program designed to improve our understanding and treatment of patients with congenital anomalies and pregnancy complications.
Facebook: http://facebook.com/UCSFcenterformaternalfetalprecisionmedicine.
Email: mfprecision@ucsf.edu.
Web site: http://mfprecision.ucsf.edu. - The Red Blood Cell Laboratory (the reference hemoglobinopathy diagnostic lab) at UCSF Benioff Children's Hospital Oakland.
- Vichinsky EP. Alpha thalassemia major--new mutations, intrauterine management, and outcomes. Hematology Am Soc Hematol Educ Program 2009:35-41. doi: 10.1182/asheducation-2009.1.35.
- Vichinsky EP. Clinical manifestations of Alpha-thalassemia. Cold Spring Harb Perspect Med 2013;3 5:a011742.
- Thalassemia.com Alpha Thalassemia.
- Derderian SC, Jeanty C, Walters MC, Vichinsky E, MacKenzie TC. In utero hematopoietic cell transplantation for hemoglobinopathies. Front. Pharmacol., 12 January 2015.
- "In-Utero HSC Transplant with Fetal Transfusion Could Lead to Cures for Many Blood Diseases", UCSF Pediatric Focus, September 2017.
- Alpha Thalassemia Major brochure for patients.
- Alpha Thalassemia Major brochure for providers.
Updated 4/10/2021



