








 Thalassemia is a genetic blood disorder.
People with Thalassemia disease are not able to make enough hemoglobin, which causes severe anemia.
Hemoglobin is found in red blood cells and carries oxygen to all parts of the body.
When there is not enough hemoglobin in the red blood cells, oxygen cannot get to all
parts of the body. Organs then become starved for oxygen and
are unable to function properly.
Thalassemia is a genetic blood disorder.
People with Thalassemia disease are not able to make enough hemoglobin, which causes severe anemia.
Hemoglobin is found in red blood cells and carries oxygen to all parts of the body.
When there is not enough hemoglobin in the red blood cells, oxygen cannot get to all
parts of the body. Organs then become starved for oxygen and
are unable to function properly.
There are two primary types of Thalassemia disease: Alpha Thalassemia disease and Beta Thalassemia disease. Beta Thalassemia Major (also called Cooley's Anemia) is a serious illness. Symptoms appear in the first two years of life and include paleness of the skin, poor appetite, irritability, and failure to grow. Proper treatment includes routine blood transfusions and other therapies.
There are two main types of Alpha Thalassemia disease. Alpha Thalassemia Major is a very serious disease in which severe anemia begins even before birth. Pregnant women carrying affected fetuses are themselves at risk for serious pregnancy and delivery complications. Another type of Alpha Thalassemia is Hemoglobin H disease. There are varying degrees of Hemoglobin H disease.
Thalassemia is a complex group of diseases that are relatively rare in the United States but common in Mediterranean regions and South and Southeast Asia. Worldwide, there are 350,000 births per year with serious hemoglobinopathies (blood disorders). In the United States, as a consequence of immigration patterns, occurrence of thalassemia disorders is increasing.
The thalassemias are a diverse group of genetic blood diseases characterized by absent or decreased production of normal hemoglobin, resulting in a microcytic anemia of varying degree. The thalassemias have a distribution concomitant with areas where P. falciparum malaria is common. The alpha thalassemias are concentrated in Southeast Asia, Malaysia, and southern China. The beta thalassemias are seen primarily in the areas surrounding Mediterranean Sea, Africa and Southeast Asia. Due to global migration patterns, there has been an increase in the incidence of thalassemia in North America in the last ten years, primarily due to immigration from Southeast Asia.
In the normal adult, hemoglobin A, which is composed of two alpha and two beta globins (A2Β2), is the most prevalent, comprising about 95% of all hemoglobin. Two minor hemoglobins also occur: hemoglobin A2, composed of two alpha and two delta globins (α2 δ2) comprises 2-3.5% of hemoglobin, while hemoglobin F, composed of two alpha and two gamma globins (α2 γ2) comprises less than 2% of hemoglobin.
Hemoglobin F, or fetal hemoglobin, is produced by the fetus in utereo and until about 48 weeks after birth. Hemoglobin F has a high oxygen-affinity in order to attract oxygen from maternal blood and deliver it to the fetus. After birth, the production of adult hemoglobin rapidly increases and fetal hemoglobin production drops off.
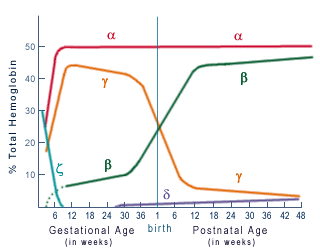 The genes controlling globin production are on chromosome 16
(alpha globin genes: "α"),
and chromosome 11
(beta: "β", gamma: "γ", and delta: "δ" genes).
As seen in the diagram, the alpha globin molecule concentration
is rather stable in fetal and adult life, because it is needed for both
fetal and adult hemoglobin production. The beta globin appears early in
fetal life at low levels and begins to rapidly increase after 30 weeks
gestational age, reaching a maximum about 30 weeks postnatally. The gamma
globin molecule reaches a high level early in fetal life at about 6 weeks
and begins to decline about 30 weeks gestational age, reaching a low level
about 48 weeks postgestational age. The delta globin appears at a low
level at about 30 weeks gestational age and maintains a low profile throughout
life.
The genes controlling globin production are on chromosome 16
(alpha globin genes: "α"),
and chromosome 11
(beta: "β", gamma: "γ", and delta: "δ" genes).
As seen in the diagram, the alpha globin molecule concentration
is rather stable in fetal and adult life, because it is needed for both
fetal and adult hemoglobin production. The beta globin appears early in
fetal life at low levels and begins to rapidly increase after 30 weeks
gestational age, reaching a maximum about 30 weeks postnatally. The gamma
globin molecule reaches a high level early in fetal life at about 6 weeks
and begins to decline about 30 weeks gestational age, reaching a low level
about 48 weeks postgestational age. The delta globin appears at a low
level at about 30 weeks gestational age and maintains a low profile throughout
life.
In the thalassemia patient, a mutation or deletion of the genes that control globin production occurs. This leads to a decreased production of the corresponding globin chains and an abnormal hemoglobin ratio (α:non-α). This abnormal ratio leads to decreased synthesis of hemoglobin and the expression of thalassemia. The globin that is produced in normal amounts winds up in excess and forms red cell aggregates or inclusions. These aggregates become oxidized and damage the cell membrane, leading either to hemolysis, ineffective erythropoiesis, or both. The quantity and properties of these globin chain aggregates determine the characterstics and severity of the thalassemia.
Beta thalassemia results in an excess of alpha globins, which leads to the formation of alpha globin tetramers (α4) that accumulate in the erythroblast (immature red blood cell). These aggregates are very insoluble and precipitation interferes with erythropoiesis, cell maturation and cell membrane function, leading to ineffective erythropoiesis and anemia.
Alpha thalassemia results in an excess of beta globins, which leads to the formation of beta globin tetramers (β4) called hemoglobin H. These tetramers are more stable and soluble, but under special circumstances can lead to hemolysis, generally shortening the life span of the red cell. Conditions of oxidant stress cause Hgb H to precipitate, interfering with membrane function and leading to red cell breakage. Hemoglobin H-Constant Spring disease is a more severe form of this hemolytic disorder. The most severe thalassemia is alpha thalassemia major, in which a fetus produces no alpha globins, which is generally incompatible with life.



